Utilisation systémique de la morphine
Sommaire
- Le traitement de la douleur périopératoire chez le chien et le chat représente un véritable défi pour les vétérinaires.
- Treatment of perioperative pain in domestic animals represents a real challenge for veterinarians.
Auteur : Dr. C. Bille 11-11-2008
Centre Hospitalier Vétérinaire des Cordeliers, 29-35 avenue du Maréchal Joffre, 77100 Meaux.
E-mail : cbille@chvcordeliers.com
Mots clefs : Opioïdes – Périopératoire – Douleur – Chien – Chat – Opioids – Perioperative – Pain – Dog – Cat
Cet article a été publié dans : Elsevier (2008) 43, p 97-17
L’utilisation systémique de la morphine et de ses
dérivés dans la gestion de la douleur périopératoire
chez le chien et le chat.
Morphine and its derivates systemic administration in the treatment of
perioperative pain in dogs and cats.
Résumé
Le traitement de la douleur périopératoire chez le chien et le chat représente un véritable défi pour les vétérinaires. Les raisons les plus fréquemment invoquées pour la non-utilisation des opioïdes sont la crainte des effets secondaires, la difficulté à identifier la douleur de manière certaine, ainsi que la lourdeur administrative imposée par l’utilisation de certains principes actifs.
La nociception comprend quatre étapes physiologiques : la transduction, la transmission, la modulation et la perception.
Ces quatre étapes sont le siège des modulations du message douloureux. Il peut être exacerbé par des phénomènes d’hypersensibilité, centrale et périphérique, de facilitation cumulative (wind up) et d’allodynie. Il peut aussi être diminué par l’utilisation d’analgésiques. La morphine et ses dérivés agissent sur les récepteurs opioïdes µ, κ et δ. La large distribution de ces récepteurs fait des opioïdes des molécules de choix dans le traitement de la douleur car ils agissent sur toutes les étapes de la modulation du message nociceptif.
En France, à ce jour, les douleurs sévères pourront être prises en charge par la morphine et le fentanyl.
Le tramadol, la péthidine, le butorphanol et la buprénorphine sont efficaces pour traiter les douleurs légères et modérées mais ne peuvent être recommandées pour le traitement des douleurs sévères.
Enfin, la naloxone peut avoir un rôle d’antagoniste sur les effets des opioïdes lorsqu’ils sont indésirables.
Summary
Treatment of perioperative pain in domestic animals represents a real challenge for veterinarians. Most frequent reasons for not giving opioids are the fear of undesirable side effects, the difficulties encountered in identifying clinical pain in companion animals and administrative requirements for some of the these molecules.
Nociception can be divided in four key steps: transduction, transmission, modulation and perception.
Modulation of the pain message takes place on one or more of these steps. It can be exacerbated by central or peripheral sensitization, wind up phenomenon or allodynia. On the other hand, it can be blunt by the use of analgesics. Morphine and its derivates act on opioid receptors µ, κ and δ. The wide distribution of these receptors makes the opioids the corner stone of pain management because they can act on every modulation step of the nociceptive message.
In France, to date, morphine and fentanyl can be used to treat severe pain.
Tramadol, meperidine, butorphanol and buprenorphine are effective to treat mild and moderate pain. Their use in the treatment of severe pain cannot be recommended.
Finally, naloxone is to be used to reverse opioid effects when they are undesirable.
Introduction
L’Association internationale pour l’étude de la douleur (IASP) définit la douleur comme « une expérience sensorielle et émotionnelle désagréable liée à des lésions tissulaires réelles ou potentielles ou décrites en termes de telles lésions » 1, 2. Il est intéressant de noter qu’une composante émotionnelle, comme l’anxiété ressentie lors d’une hospitalisation, peut participer à la sensation de douleur.
La douleur n’a pas toujours occupé le premier plan dans la thérapeutique vétérinaire.
Dans une étude menée au sein de l’université de Caroline du Nord, aux États-Unis, dans les années 1980, portant sur 243 chiens ayant subi des amputations, des greffes osseuses appendiculaires (limb sparing), des thoracotomies, des traitements d’instabilité cervicale et des réparations de fracture fibulaire, seuls 40 % des animaux traités ont reçu un analgésique, quel qu’il soit, après l’intervention chirurgicale 3.
En 1987, Crane 4 exprime l’idée selon laquelle, chez le chien, une douleur modérée provoquée par une hernie discale est mieux traitée par le confinement et la mise au repos forcée (cagéothérapie) que par l’administration d’analgésiques. L’animal est considéré comme « bénéficiant » de la douleur qu’il ressent. Cette idée est encore largement répandue de nos jours.
En 1996, au Canada, 275 vétérinaires ont été questionnés sur l’utilisation qu’ils faisaient des opioïdes lors de chirurgies abdominales, orthopédiques avec pose de fixateurs, du ligament croisé, de convenance et d’onychectomie. Moins de 50 % des confrères interrogés ont été considérés comme des utilisateurs réguliers 5, 6.
En 1999, au Royaume-Uni, d’après les réponses de 958 confrères, seuls 53 % des animaux subissant une ovariohystérectomie et 71 % des animaux subissant une chirurgie abdominale ont reçu un analgésique (opioïde ou antiinflammatoire).
Un tiers des chirurgiens considéraient qu’« un degré de douleur [était] nécessaire pour limiter l’activité de l’animal après la chirurgie » et 6,5 % que « l’intervention chirurgicale considérée ne [provoquait] pas suffisamment de douleur pour qu’elle justifie la mise en place d’un traitement » 7.
En 2004, en France, d’après une enquête menée auprès de 189 confrères, il apparaît que les opioïdes ne sont utilisés que chez 16,1 % des chiens et 8,1 % des chats, toutes causes de douleur confondues. Seuls 17,2 % des chiens et 36,3 % des chiennes stérilisés ont rec¸u un analgésique (antiinflammatoire ou opioïde) 8.
Les raisons les plus fréquemment invoquées pour la nonutilisation de dérivés morphiniques sont les mêmes, quel que soit le pays considéré. Il s’agit des risques de dépression respiratoire, de bradycardie et de sédation. De plus, les vétérinaires expriment leur difficulté à identifier et quantifier un état algique. Enfin, la législation semble freiner l’acquisition et l’utilisation de substances narcotiques 5-8.
Le traitement de la douleur, s’il a longtemps été délaissé, est désormais mieux pris en considération. Tout d’abord, une meilleure compréhension des mécanismes mis en jeu a permis d’établir que la douleur a des conséquences physiologiques néfastes. Elle provoque une activation du tonus orthosympathique responsable de la libération de catécholamines. Le débit cardiaque, les résistances vasculaires périphériques, la pression artérielle et la consommation en oxygène par le myocarde sont augmentés. La sécrétion de certaines hormones cataboliques (ACTH, cortisol, glucagon, ADH, hormone de croissance. . .) est stimulée, tandis que celle des hormones anaboliques (insuline, testostérone. . .) est inhibée.
La balance métabolique est modifiée et l’animal est placé dans un « état catabolique » qui limite sa guérison 2, 9, 10.
De plus, la mise en place d’un plan d’analgésie efficace et précoce permet de diminuer les doses d’anesthésiques utilisés et donc de limiter les dépressions cardiovasculaire et respiratoire dont ils peuvent être à l’origine.
Enfin, de nouvelles molécules analgésiques sont disponibles sur le marché vétérinaire. Aux États-Unis, la Joint Commission on Accreditation of Healthcare Organisation a défini en 2000 la douleur comme le cinquième paramètre vital chez le patient humain après le pouls, la respiration, la température et la pression artérielle. De même, l’American Animal Hospital Association recommande aux vétérinaires de considérer la douleur comme le quatrième signe vital après le pouls, la respiration et la température 2.
Quel que soit le type de douleur, elle est néfaste pour l’animal et toutes les mesures nécessaires doivent être mises en place pour la traiter. Elle est parfois prévisible (douleur chirurgicale) et doit être traitée préventivement : c’est le concept de l’analgésie préventive 2, 11-13.
Cet article a pour objectif de faire le point sur l’utilisation systémique des différents types de dérivés morphiniques dont les praticiens disposent lorsqu’ils réalisent une intervention chirurgicale, même mineure. La première partie sera consacrée à un bref rappel de la physiologie de la douleur. Puis, l’ensemble des analgésiques morphiniques disponibles sur le marché sera exposé, afin que les vétérinaires puissent choisir le plus approprié à leur pratique courante.
Physiologie de la douleur
De la stimulation à la sensation de douleur
La nociception comprend quatre étapes physiologiques : la transduction, la transmission, la modulation et la perception. Elle met en jeu les fibres périphériques, les voies spinales ascendantes et les systèmes d’intégration et de modulation du message nerveux 1.
Les terminaisons nerveuses des fibres périphériques sont sensibles à des stimulations mécaniques, chimiques ou thermiques. Ce sont les nocicepteurs. Ils transmettent les messages par deux types de fibres. Les fibres Aδ, d’une part, qui sont responsables de la douleur immédiate. La transmission par ces fibres est rapide, aiguë, localisée et se termine avec la cessation de la stimulation. Les fibres C, d’autre part, sont responsables d’une douleur plus tardive. La transmission par ces fibres est plus lente, diffuse et persistante 1, 14.
Le premier relais synaptique a lieu dans la substance grise de la moelle épinière, dans la corne dorsale. Les fibres Aδ et C participent à des relais avec trois types de neurones, des interneurones qui permettent une modulation locale du message, des fibres proprioceptives qui participent à l’arc réflexe, des fibres des voies ascendantes qui transmettent le message aux centres supraspinaux 1.
Les voies ascendantes transmettent le message aux centres intégrateurs (thalamus, hypothalamus, formation réticulée) par les voies spinothalamique, spinoréticulée ou trigéminale 1, 2, 10.
Le message nerveux, sur les centres intégrateurs, a pour effet de moduler l’homéostasie, de provoquer des réponses motrices et des réponses affectives ou de modifier le comportement 1.
Modulation du message nociceptif
Des mécanismes régulateurs, inhibiteurs ou amplificateurs du message nociceptif, sont en permanence mis en place par l’organisme. Nous les considérerons indépendamment les uns des autres et les classifierons selon leur principale localisation anatomique.
Modulation périphérique
La libération de certaines substances (médiateurs de l’inflammation, potassium, adénosine triphosphate, substance P. . .) dans l’environnement des récepteurs périphériques modifie le message douloureux. Elle peut provoquer une augmentation de la réactivité des nocicepteurs à une stimulation douloureuse : on parle de sensibilisation périphérique 1, 2, 10, 14. La douleur ressentie lorsqu’on se coince la main dans une porte est beaucoup plus vive si celle-ci est déjà blessée.
Corne dorsale
Il reste de nombreux points non élucidés concernant la modulation du message nociceptif dans la corne dorsale. Il semblerait qu’une population neuronale (les neurones wide dynamic range) localisée dans cette région, soumise à l’action du neuromédiateur N-méthyl-d-aspartate (NMDA), soit en partie responsable du phénomène de facilitation cumulative de la réponse (wind up) 12, 13, 15.
Cette facilitation correspond au recrutement d’un grand nombre de neurones postsynaptiques dans la corne dorsale en réponse à un stimulus nociceptif de forte intensité appliqué pendant un période prolongée à un récepteur périphérique.
Cette facilitation a plusieurs conséquences. Elle augmente la sensibilité des neurones environnants dans la corne dorsale. Cela sensibilise les tissus périphériques sains qui sont dans l’environnement direct de la zone lésée : c’est le phénomène de sensibilisation centrale. Elle peut être à l’origine de la perception douloureuse d’un message qui ne l’est pas dans des conditions habituelles : on parle d’allodynie 1, 2, 10. Classiquement, il s’agit de la douleur que l’on ressent lorsque l’on caresse un coup de soleil.
L’une des particularités du wind up est qu’il n’est pas aboli par l’anesthésie générale.
Centres intégrateurs
Plusieurs régions de l’encéphale (thalamus, cortex, système limbique. . .) sont activées par les messages douloureux. Elles sont responsables de la libération de neurotransmetteurs (endorphines, enképhalines, dynorphine, dopamine. . .) qui permettent la modulation du message nociceptif 1, 2. C’est lors de cette étape que l’excitation, l’anxiété ou la peur du patient peut modifier sa perception et exacerber le ressenti de la douleur.
Différents « types » de douleur
Plusieurs concepts ont été proposés pour classer la douleur. Aucun n’est complètement satisfaisant. Ces concepts permettent cependant d’appréhender certaines notions en classant la douleur en plusieurs « types ».
Douleurs adaptatives et non adaptatives
Si, traditionnellement, on distinguait une douleur aiguë et une douleur chronique, en se basant sur le temps d’évolution 1, 14, la tendance est aujourd’hui de classer la douleur comme adaptative ou non adaptative 2. La douleur adaptative est définie comme une réponse « normale » de l’organisme à des dégâts tissulaires, comme un traumatisme, un geste chirurgical ou une infection.
Elle décroît et disparaît lorsqu’un traitement étiologique est mis en place. Si la douleur adaptative n’est pas traitée, elle peut être modulée par les centres intégrateurs et devenir non adaptative. C’est le cas d’une douleur qui évolue au-delà du temps nécessaire à une plaie ou une infection pour qu’elle guérisse. Elle peut aussi être associée à des affections chroniques comme l’arthrose ou certains cancers 2.
Douleur neuropathique
La douleur neuropathique est provoquée par une lésion du système nerveux central ou périphérique. Si tous les mécanismes mis en jeu sont à ce jour mal compris, il semble admis qu’elle est à l’origine d’une modification des processus de transmission du message douloureux.
Une amputation, un traumatisme, une thromboembolie ou une endocrinopathie peuvent en être à l’origine. La douleur neuropathique est frustrante pour le praticien dans ce sens qu’elle répond généralement mal aux administrations d’analgésiques (anti-inflammatoires, opioïdes).
Il semblerait que, dans la corne dorsale, le NMDA joue un rôle prépondérant par l’effet de facilitation cumulative (wind up) 1, 2, 12, 13, 15, 16. Ainsi, certaines molécules antagonistes du récepteur NMDA, la kétamine notamment, semblent améliorer les dysfonctionnements de la transmission et des contrôles des messages douloureux 12, 13, 15, 16.
Douleur viscérale
La douleur viscérale est provoquée par l’atteinte d’un organe. Le plus souvent, il s’agit d’une maladie et non d’une blessure. Les terminaisons nerveuses des viscères sont plus sensibles à la distension, à l’ischémie et à l’inflammation qu’aux stimulations thermiques ou chimiques. Les phénomènes d’allodynie ou de facilitation cumulative n’ont pas été précisément décrits comme associés à des douleurs viscérales 1.
Analgésiques morphiniques disponibles en pratique courante
Opioïdes : définition et mode d’action
Les opioïdes se définissent comme des dérivés de l’opium, naturels ou de synthèse. Si leurs mécanismes d’action ne sont pas entièrement élucidés, il est admis qu’ils agissent par leurs interactions avec au moins trois types de récepteurs : µ, κ et δ. Sur chaque récepteur, le principe actif va avoir un effet activateur (effet agoniste) ou inhibiteur (effet antagoniste). La Fig. 1 présente les différentes interactions possibles entre un opioïde et son récepteur. La stimulation de chaque récepteur va induire une réponse différente. On parle d’effet µ, κ et δ. À ce jour, il n’a pas été possible d’isoler et de définir avec précision ces effets. Tout d’abord, parce que cette classification, pratique pour la compréhension, est probablement abusive. Un quatrième type de récepteur a été identifié : le récepteur à la nocicéptine 17. Ensuite, parce que différents sous-types de récepteurs ont été décrits : les µ1, µ2, µ3, κ1a, κ1b, κ2, κ3, δ1 et δ2 17 et qu’il n’existe pas de principe actif qui n’agisse que sur un seul type ou un seul sous-type de récepteur 18-20.
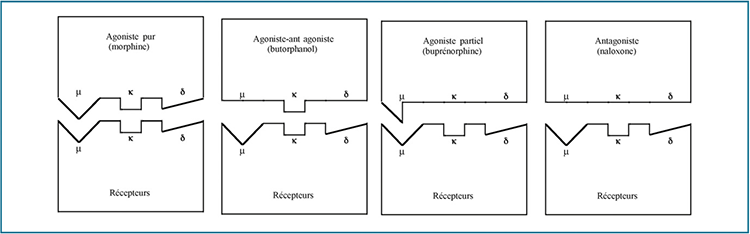
Figure 1. Modélisation des interactions entre les opioïdes et leurs récepteurs.
Les récepteurs µ, κ et δ sont répartis dans tout l’organisme. Il en découle deux notions qui sont à la base de l’utilisation des opioïdes. En premier lieu, considérant que le pouvoir analgésique d’un opioïde résulte de son effet agoniste sur les récepteurs µ et/ou κ 17, 21, cette large distribution en fait des molécules qui interviennent sur toutes les étapes de la transmission du message nociceptif (récepteurs périphériques, voies ascendantes, intégration et voies descendantes). À titre d’exemple, l’administration intraarticulaire de morphine permet d’obtenir une analgésie satisfaisante chez les chiens subissant une arthrotomie 22, 23. Elle semble agir sur des récepteurs opioïdes périphériques de type µ 24. Dans la corne dorsale, les opioïdes inhibent la transmission synaptique du message 21. Dans les centres intégrateurs, ils activent les mécanismes inhibiteurs descendants et agissent sur le système limbique en diminuant la composante émotionnelle de réponse à la douleur 21.
De plus, cette distribution des récepteurs aux opioïdes dans de nombreux organes permet d’expliquer et d’appréhender la diversité de leurs effets secondaires.
Nous ne décrirons que ceux qui sont impliqués dans la gestion de la période périopératoire. Les opioïdes, par leur action sur les récepteurs µ, ont un effet dépresseur sur le système nerveux central 21. On observe un effet dépresseur respiratoire qui résulte de la perte de sensibilité des centres de la respiration à l’augmentation de la concentration plasmatique de CO2 21. Les centres de la thermorégulation et de la toux sont aussi inhibés 21. À l’inverse, la chemoreceptive trigger zone (CTZ) est stimulée. Cette stimulation est responsable des vomissements que l’on observe dans certains cas 21. Chez le chat, il est possible d’observer un effet stimulateur du système nerveux central responsable d’une hyperexcitation 17. L’augmentation du tonus parasympathique provoque une bradycardie dose-dépendante 21. Celle-ci peut être antagonisée par l’administration d’un anticholinergique (atropine ou glycopyrrolate). Les opioïdes, par l’association de leurs effets centraux et périphériques, provoquent une augmentation des contractions intestinales segmentaires, associée à une inhibition du complexe moteur migrant, donc du péristaltisme. Il peut en résulter une constipation 21. Ils pourraient être à l’origine de spasmes des sphincters des voies biliaires et pancréatiques 21. Enfin, la stimulation des récepteurs µ favorise la synthèse d’ADH et diminue la diurèse alors que celle des récepteurs κ diminue la production d’ADH et stimule la diurèse 9, 21.
Différents types d’opioïdes
Chaque opioïde peut se définir par son action sur les différents récepteurs.
Agonistes µ
Il s’agit de la morphine, du fentanyl, du tramadol et de la mépéridine 21, 25.
- Morphine
La morphine est la molécule de référence. C’est un agoniste µ, κ et δ. Elle est disponible sous forme injectable (0,1, 1, 10, 20 ou 40 mg/mL) ou orale à libération prolongée (Skenan® 10, 20, 30, 60, 100 et 200 mg ou Kapanol® 20, 50, 100 mg) dans les pharmacies d’officine. Son obtention nécessite l’utilisation d’ordonnances sécurisées. Elle peut être administrée par voie intraveineuse (i.v.) lente, intramusculaire (i.m.), sous-cutanée (s.c.) ou orale (per os : p.o.).
Chez le chien, la dose, par voie injectable, est comprise entre 0,1 et 0,5 mg/kg 13, 25, 26. La dose de la forme orale est comprise entre 1,5 et 3 mg/kg toutes les 12 heures 17.
Chez le chat, seule la voie injectable est utilisée 17. La dose est comprise entre 0,05 et 0,2 mg/kg 2, 27.
Son délai d’action, lorsque la voie i.v. lente est utilisée, est d’environ cinq minutes. Il est d’environ 15 minutes pour la voie i.m., 30 minutes pour la voie s.c. et une heure pour la voie orale.
Sa demi-vie d’élimination est d’environ 90 minutes 28. Son effet analgésique dure en moyenne trois ou quatre heures pour les formes injectables et environ 12 heures pour les formes orales à libération prolongée 11, 17, 25.
Chez le chien et le chat, l’utilisation de morphine permet de diminuer la quantité d’isoflurane nécessaire pour abolir les réponses motrices aux stimulations nociceptives 12, 29. Cependant, chaque animal réagit différemment et l’effet analgésique des opioïdes doit être régulièrement évalué, afin de s’assurer du bon contrôle de la douleur. Il semblerait que l’administration de morphine en continu, par perfusion, soit très efficace pour contrôler la douleur 12, 30. Une étude portant sur 20 chiens subissant une laparotomie a comparé les effets de l’administration de morphine par perfusion, à la dose de 0,12 mg/kg par heure, versus par voie i.m., à la dose de 1,1 mg/kg toutes les quatre heures sur 24 heures. Aucune différence significative n’a été observée concernant l’état algique des animaux entre les deux groupes. Cependant, l’utilisation de morphine par perfusion permet de diviser de moitié la dose de morphine totale nécessaire sur 24 heures 30. La dose recommandée chez le chat et le chien est de 0,1 à 2 mg/kg par heure 11—13, 25, 29, 30.
Chez le chien, l’injection de morphine par voie i.v. pourrait provoquer un relargage histaminique pouvant mener à une hypotension. L’intensité de ce dernier dépendrait de la rapidité d’injection 25. Aussi, il est conseillé d’administrer la morphine par voie i.v. lente, sur environ une minute. L’utilisation de morphine sur des animaux anesthésiés peut provoquer un reflux gastro-oesophagien et donc être contre-indiquée pour certains types d’interventions chirurgicales. Il semble que cet effet soit dose-dépendant. Une étude portant sur 90 chiens a montré que 27 % des animaux n’ayant pas rec¸u de morphine, 50 % de ceux ayant rec¸u 0,22 mg/kg en i.m. et 60 % de ceux ayant reçu 1,1 mg/kg en i.m. ont présenté un reflux gastro-œsophagien 31. - Fentanyl
Le fentanyl est un opioïde de synthèse agoniste µ. Certains considèrent son pouvoir analgésique comme étant 80 à 150 fois plus puissant que celui de la morphine 25, 32. Il peut être administré par voie i.v. ou transdermique. Il est disponible en pratique vétérinaire sous sa forme injectable à 5 % depuis environ 18 mois. Son obtention se fait directement auprès des laboratoires AguettantTM, MerckTM, PanpharmaTM, Dakota PharmTM, RenaudinTM ou JanssenTM. Il est aussi disponible, en pharmacie d’officine, sous forme de patchs transdermiques (Durogesic®) qui diffèrent par leur débit d’administration : 25, 50, 75 et 100 µg par heure. L’obtention de fentanyl, quel que soit son conditionnement, nécessite l’utilisation d’ordonnances sécurisées.
C’est une molécule lipophile qui est rapidement redistribuée dans les tissus. Son action est plus rapide et plus brève que celle de la morphine. Après une injection de fentanyl par voie i.v., la concentration plasmatique de la molécule permettant d’obtenir une analgésie est atteinte en 2,5 minutes 32. Sa demi-vie d’élimination est de 45 minutes 32. Son effet clinique ne dure que 20 à 30 minutes 25. Cela impose donc une utilisation sous forme de perfusion, après l’administration d’un bolus de charge.
Chez le chien, la dose de charge est comprise entre 2 et 5µg/kg en bolus, suivie d’une perfusion comprise entre 2 et 10µg/kg par heure 9, 25, 32. Chez le chat, il est conseillé d’utiliser une dose de charge comprise entre 2 et 3 µg/kg en i.v. et une perfusion comprise entre 0,5 et 2,5 µg/kg par heure 9, 25, 33. Certains l’utilisent chez le chien en administrant des bolus successifs de 2,5 µg/kg toutes les 30 minutes 34.
Le fentanyl diminue significativement les quantités d’enflurane et d’isoflurane nécessaires pour abolir les réponses motrices aux stimulations nociceptives 35—37.
Chez le chien et le chat, pour différentes indications (laparoscopie, chirurgie orthopédique, onychectomie, stérilisation), le fentanyl est efficace pour traiter la douleur 34, 38—42.
Les dispositifs d’administration transdermique (patchs) sont disponibles pour quatre débits d’administration : 25, 50, 75 et 100 µg par heure 11, 25. Les doses recommandées sont les suivantes :- un chat de plus de 2,3 kg et un chien de 3 à 10kg: patch de 25µg par heure ;
- un chien de 10 à 20 kg : patch de 50 µg par heure ;
- un chien de 20 à 30 kg : patch de 75 µg par heure ;
- un chien de plus de 30 kg : patch de 100 µg par heure 25, 43.
La concentration plasmatique effective de fentanyl est atteinte en 24 heures et reste stable pendant 48 heures (72 heures après la pose du dispositif) 25, 44. Plusieurs études ont prouvé l’efficacité de ce dispositif chez le chien et le chat dans le contrôle de la douleur provoquée par une chirurgie orthopédique, une onychectomie ou une ovariectomie 38—42, 45. Cependant, cette voie d’administration peut être décevante pour contrôler une douleur opératoire. La molécule active n’est dans le sang en concentration suffisante que 24 heures après la pose du dispositif 25, 44. Cela impose de voir l’animal la veille de l’intervention et interdit l’utilisation de ce dispositif sur toute intervention non prévue. La dose de molécule active effectivement absorbée est extrêmement variable d’un animal à l’autre 44, 45.
Il semble que le lieu d’application du patch (encolure, thorax, abdomen) induise des variations dans la quantité de produit absorbé 25, 46. L’hypothermie, souvent présente en période périopératoire, diminue significativement l’absorption transdermique de fentanyl 37, 47. La préparation de la peau est aussi à l’origine des variations d’absorption (utilisation de tondeuse, rasoir, crème dépilatoire, savon, alcool. . .). Ainsi, il est conseillé de préparer le lieu d’application par une tonte, les poils et d’éviter toute autre manipulation, comme l’épilation, le savonnage
ou l’application d’alcool 25, 48. L’utilisation d’un dispositif d’administration transdermique ne simplifie pas la tâche de l’équipe médicale.
Une étude a comparé deux groupes de chiens. L’un recevait de la morphine par voie i.m. toutes les quatre heures et l’autre, du fentanyl par voie transdermique. Les deux groupes ont nécessité la même surveillance postopératoire. Le taux de recours à un analgésique d’urgence était le même dans les deux groupes. Les dépenses financières engagées sur les animaux recevant du fentanyl étaient plus importantes 40. Selon les recommandations usuelles, ce dispositif oblige le praticien à traiter un animal de 11 kg avec la même dose qu’un animal de 19 kg. Il est impossible qu’ils reçoivent chacun une dose efficace de fentanyl avec le même patch. Soit l’un est surdosé, soit l’autre est sousdosé.
Il est formellement déconseillé de couper le patch car cela détruit la membrane alvéolée qui règle le débit d’administration et le produit peut se rependre sur la peau par le bord de coupe.
L’animal court alors un risque de surdosage. Il peut éventuellement être conseillé de recouvrir la moitié de la surface de contact avec la peau de manière à diminuer le débit. Ce dispositif interdit toute adaptation de la posologie sur des chiens qui ressentent des douleurs aiguës peropératoires. Enfin, aux États-Unis, la Food and Drug Administration a retiré son autorisation de mise sur le marché au patch de fentanyl pour le traitement des douleurs aiguës postopératoires chez les patients humains 40.
- Tramadol
Le tramadol est un opioïde de synthèse qui possède un faible effet agoniste µ. Il est disponible sous forme injectable (Contramal®, Topalgic®, 100 mg/2 mL) ou orale (tramadol générique 50 mg) dans les pharmacies d’officine. Son obtention ne nécessite pas l’utilisation d’ordonnances sécurisées. En plus de son effet agoniste µ, il empêche aussi la recapture neuronale de noradrénaline et de sérotonine 15, 26, 49, 50.
Il est supposé que cet effet antisérotoninergique joue un rôle dans son pouvoir analgésique. Le tramadol est actif sous deux formes : le tramadol lui-même et la molécule M1 issue de sa métabolisation. La molécule M1 possède une affinité 200 fois supérieure pour les récepteurs µ que le tramadol. Son délai d’action chez les animaux de compagnie et sa demi-vie d’élimination n’ont pas été étudiés. Cependant, l’effet clinique est généralement observé dans les deux heures qui suivent son administration et dure environ six heures.
Chez le chien et le chat, la dose est de 1 à 4 mg/kg, i.v., p.o. 49—51. Sur des chiens subissant une intervention chirurgicale provoquant une douleur modérée (ovario-hystérectomie), il n’a pas été observé de différence significative entre un groupe ayant rec¸u du tramadol et un groupe ayant rec¸u de la morphine 49.
Chez le chien et le chat, le tramadol semble efficace pour la prise en charge des douleurs modérées, mais n’est pas indiqué dans le traitement des douleurs sévères 15, 26, 49, 50. Son efficacité est discutée chez le chat 51,52. Son utilisation pourrait être intéressante dans le traitement de l’allodynie et de l’hyperalgie provoquée par la sensibilisation centrale ou périphérique 15.
Les effets secondaires observés chez l’humain sont un état nauséeux, une constipation, des maux de tête, une somnolence, des vomissements, du prurit, une stimulation du système nerveux central, une asthénie, une sécheresse buccale et de la diarrhée 50.
Le tramadol est métabolisé par le foie. Il subit notamment une glucuronoconjugaison. Il est contre-indiqué en cas de troubles hépatiques 50.
Son effet sur la concentration de sérotonine contre-indique cette molécule lorsque les animaux rec¸oivent des traitements antidépresseurs, comme la séléginine, ou présentent des antécédents de crises convulsives 50.
- Mépéridine ou péthidine
La péthidine est un opioïde de synthèse qui possède un faible effet agoniste µ, κ et δ. Elle a aussi pour effet de bloquer les canaux à sodium et l’activité de la corne dorsale comme un anesthésique local (lidocaïne, bupivacaïne. . .). Enfin, il semble qu’elle exerce un effet α2 agoniste comme la dexmédétomidine, la médétomidine, la romifine ou la xylazine 11. La péthidine est produite sous sa forme injectable (100 mg/2 mL) par le laboratoire RenaudinTM. Son obtention nécessite l’utilisation d’ordonnances sécurisées.
Sa dose chez le chien et le chat est comprise entre 4 et 8,8 mg/kg 11, 25, 26, 53—55. La péthidine ne s’administre que par voie i.m. ou s.c. car son utilisation i.v., même lente, peut déclencher un choc anaphylactique 25, 26, 55. Sa durée d’action est comprise entre 30 minutes et deux heures 11, 25, 54. À l’inverse des autres opioïdes, la péthidine possède un effet inotrope négatif qui contre-indique son utilisation sur les patients dont le statut cardiovasculaire est compromis. Elle exerce un effet atropine-like qui provoque une augmentation de la fréquence cardiaque 11.
La péthidine est moins efficace que la morphine pour prendre en charge les douleurs modérées. Elle n’est pas indiquée dans le traitement des douleurs sévères 53—55.
Agonistes—antagonistes
Les agonistes—antagonistes, comme le butorphanol (Dolorex®), sont des opioïdes de synthèse qui possèdent une action différente sur plusieurs récepteurs. Ils sont caractérisés par un effet µ antagoniste et un effet κ agoniste. L’effet analgésique du butorphanol découle de son interaction avec les récepteurs κ 9, 21, 25. Son effet µ antagoniste fait qu’il provoque moins d’effets secondaires indésirables (bradycardie, bradypnée, sédation) que les agonistes µ 9, 21, 25, 29, 56. Le butorphanol possède un effet plafond, c’est-à-dire que jusqu’à une concentration sanguine maximale, pour une dose administrée d’environ 0,4 mg/kg, les effets cliniques sont dose-dépendants. Lorsque cette concentration est atteinte, toute nouvelle administration ne produira plus d’effet cliniquement visible 25, 29.
Sa dose chez le chien et le chat est de 0,2 à 0,4 mg/kg. Il est administré par voie i.v., i.m. ou s.c. 29, 56. Son effet clinique apparaît en environ 30 minutes et sa durée d’action est comprise entre une et quatre heures 11,25. L’efficacité du butorphanol dans le traitement de la douleur a longtemps fait débat. En 1994, une étude menée sur six chiens n’a pas montré d’effet du butorphanol (0,2 mg/kg, 0,4 mg/kg ou 0,8 mg/kg en i.v.) sur la quantité d’halothane nécessaire pour abolir les réponses motrices aux stimulations nociceptives 57. À l’inverse, d’autres études menées chez le chien et le chat ont montré que le butorphanol (0,4 mg/kg ou 0,8 mg/kg en i.v.) diminuait significativement les quantités d’isoflurane nécessaires pour abolir les réponses motrices aux stimulations nociceptives 29, 56.
Plusieurs auteurs ont démontré que, chez le chien et le chat, pour différentes indications (laparotomie, cystotomie ou splénectomie, arthrotomie, synovite aiguë, ovario-hystérectomie, onychectomie, stérilisation), les anti-inflammatoires non stéroïdiens (kétoprofène, méloxicam, flunixine meglubine) utilisés seuls ou en association avec le butorphanol étaient plus efficaces pour traiter la douleur que le butorphanol utilisé seul 58—62.
Chez le chien, l’utilisation de butorphanol à la dose de 0,8 mg/kg s’est avérée efficace pour traiter la douleur d’origine viscérale 63.
Chez le chat, son utilisation à une dose comprise entre 0,1 et 0,4 mg/kg s’est avérée efficace pour traiter la douleur provoquée par une stimulation thermique 33, une onychectomie 64 ou une ovario-hystérectomie 65. Cependant, cet effet est de courte durée : une ou deux heures 33, 65. À ce jour, le butorphanol semble pouvoir être considéré comme satisfaisant pour traiter les douleurs modérées 29, 33, 56, 63—65. Son efficacité est accrue lorsqu’il est associé à des anti-inflammatoires non stéroïdiens ou à de la médétomidine 58—62, 65. Utilisé seul, le butorphanol n’est pas efficace pour prendre en charge des douleurs sévères 11, 25.
L’effet µ antagoniste du butorphanol peut être utilisé pour contrer les effets indésirables (bradycardie, bradypnée, sédation) provoqués par les agonistes purs (morphine, fentanyl). Son utilisation dans cette indication est très intéressante car, contrairement à celle d’un antagoniste pur comme la naloxone, elle permet de continuer à traiter la douleur par l’effet κ 9,25, 26.
Il a longtemps été considéré que l’utilisation conjointe d’agonistes purs et d’agonistes—antagonistes résultait dans une simple sommation de leurs effets. Cependant, deux études récentes ont bouleversé ces considérations.
Une étude menée par stimulation thermique sur six chats a montré que l’association de l’hydromorphone (agoniste pur) et du butorphanol avait un pouvoir analgésique inférieur à celui de l’hydromorphone utilisée seule. Cependant, l’analgésie était significativement plus longue (540 minutes versus 340 minutes) lorsque les deux molécules étaient associées 66.
À l’inverse, il a été montré, par inflation d’un ballonnet colique sur huit chats, que l’association de l’oxymorphone (agoniste pur) et du butorphanol était plus efficace dans le traitement de la douleur viscérale que l’une ou l’autre des molécules utilisées seules. Il a, de plus, été montré que les effets étaient synergiques et non simplement additifs 67.
Il semble que les interactions entre ces deux classes d’opioïdes soient bien plus complexes qu’il n’y paraît et de nouvelles recherches devront les étudier.
Agonistes partiels
La buprénorphine est un opioïde de synthèse qui possède une action agoniste partielle sur le récepteur µ et l’antagoniste sur les récepteurs κ 21, 25. Elle possède une affinité extrêmement forte pour ces récepteurs, mais n’entraîne pas une réponse clinique complète. Cela permet de limiter les effets secondaires indésirables (bradycardie, bradypnée, sédation). Elle possède aussi un effet plafond. Sa forme injectable, Temgesic® 0,3 mg/mL, est disponible auprès du laboratoire Schering-PloughTM et nécessite l’utilisation d’ordonnances sécurisées. La dose chez le chien et le chat est de 5 à 20µg/kg. L’administration en i.v. peut provoquer un relargage d’histamine. Elle ne s’administre donc que par voie i.m. Les effets cliniques sont observés une ou deux heures après l’injection et sa durée d’action est comprise entre six et 12 heures 11, 25, 26, 33. La pharmacocinétique de la molécule active est très variable d’un individu à l’autre. Cela explique que la durée d’action puisse passer du simple au double. Les éventuelles réadministrations de buprénorphine doivent être réalisées en fonction de la réponse clinique de chaque individu 68.
Différentes études chez le chien ont montré que la buprénorphine était efficace pour traiter la douleur provoquée par une arthrotomie ou une ovario-hystérectomie pendant les 24 heures qui suivent la chirurgie 69, 70.
Chez le chat, une dose de 10µg/kg est efficace pour traiter la douleur provoquée par une stimulation thermique, une onychectomie ou une ovario-hystérectomie 33, 71, 72.
Une étude portant sur 32 chats semble démontrer que, dans cette espèce, la buprénorphine, utilisée à 10 µg/kg en i.m., est plus efficace que la morphine, utilisée à 0,1 mg/kg, en i.m., pour traiter la douleur postopératoire pendant 420 minutes, qu’elle soit modérée ou sévère 73. Ces observations peuvent être dues au fait que la durée d’action de la morphine est plus courte que celle de la buprénorphine. Sur les 420 minutes d’évaluation, une partie des observations a été réalisée sur des animaux pour lesquels les effets de la morphine s’étaient dissipés.
Comme le butorphanol, la buprénorphine est considérée comme satisfaisante pour traiter les douleurs modérées. En l’état actuel des connaissances, il ne peut être conseillé de l’utiliser seule pour prendre en charge des douleurs sévères 11, 25, 33, 69, 70.
Elle peut avoir un rôle d’antagoniste sur les effets secondaires des agonistes µ 25, 26.
Sa forte affinité pour les récepteurs µ peut, dans certaines situations, poser problème. Elle est très difficile à antagoniser pendant les six à 12 heures de son action, même avec des antagonistes purs comme la naloxone.
De plus, lorsqu’elle est utilisée comme préanesthésique, elle limite le recours aux autres opioïdes, notamment les agonistes µ, car elle antagonise leurs effets par une affinité plus importante et une durée d’action plus longue 26.
Antagonistes
La naloxone est un opioïde de synthèse qui possède une forte affinité pour les récepteurs µ et κ, mais ne produit pas d’effet aux doses recommandées. Sa forte affinité pour ces derniers a pour conséquence qu’elle peut prendre la place de presque tous les autres opioïdes (naturels, synthétiques et endogènes), à l’exception de la buprénorphine, sans provoquer de réponse clinique. Il est important de noter que l’administration de naloxone antagonise tous les effets des opioïdes, y compris les effets analgésiques. Son utilisation doit être réservée aux urgences extrêmes. Son obtention (Naloxone®, Nalone®, Narcan® 0,4 mg/mL) se fait directement auprès d’une pharmacie d’officine et nécessite l’utilisation d’ordonnances sécurisées. Elle s’administre par voie i.v. La dose est de 1 à 25µg/kg. Il peut être intéressant de travailler avec les dosages les plus faibles et d’administrer plusieurs bolus successifs jusqu’à ce que l’effet souhaité soit obtenu. De cette manière, il peut être possible d’antagoniser les effets secondaires indésirables sans abolir la totalité de l’analgésie. Sa durée d’action est courte, entre 30 et 60 minutes. Son utilisation devra toujours se faire sous étroite surveillance afin qu’une renarcotisation puisse être évitée lorsque ses effets se dissipent 25.
En cas de renarcotisation, des bolus réitérés devront être administrés.
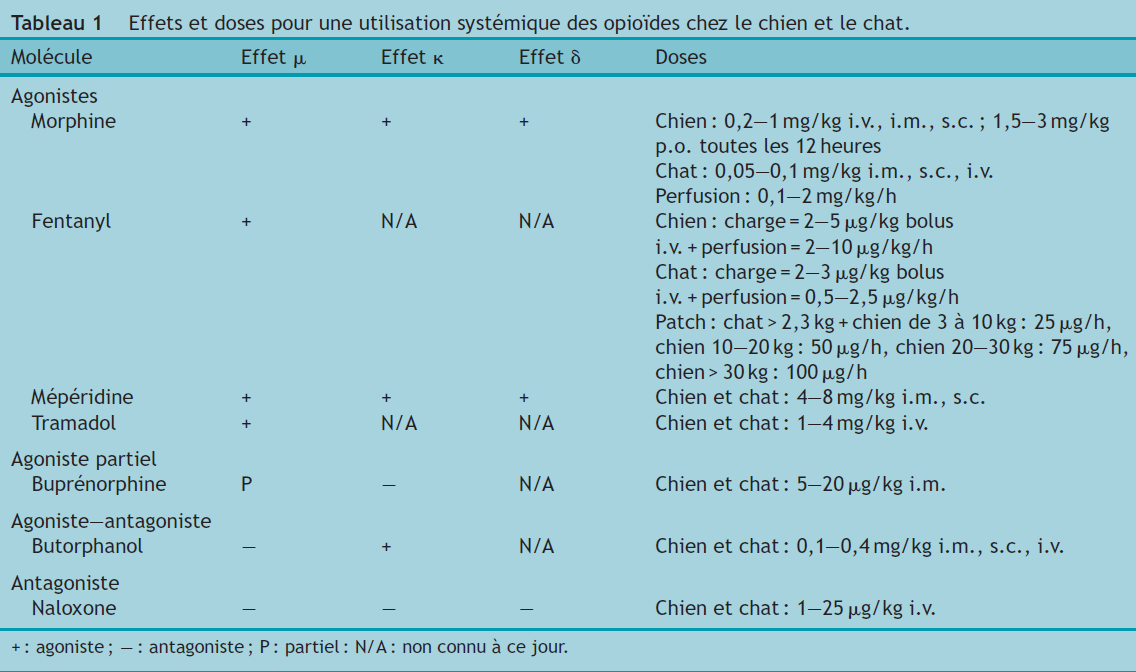
Le Tableau 1 présente une synthèse du mode d’action et des doses recommandées pour chacune des molécules présentées.
Conclusion
La compréhension des mécanismes physiologiques de la douleur, même si elle n’est pas complète, a permis de mettre en évidence certains processus clés. Ainsi, des étapes comme la facilitation cumulative (wind up), les sensibilisations centrale et périphérique ou l’hyperalgie peuvent être identifiées et spécifiquement traitées lors de l’établissement d’un plan d’analgésie.
La morphine et ses dérivés sont à ce jour les principes actifs analgésiques systémiques les plus efficaces, probablement parce qu’ils interviennent sur toutes les étapes de la propagation du message nerveux nociceptif. Il existe cependant de nombreuses situations cliniques dans lesquelles l’utilisation systémique des opioïdes seuls est insuffisante.
Le moment de l’administration d’un antalgique est crucial. Il est plus efficace s’il est administré avant la stimulation douloureuse. C’est le concept de l’analgésie préventive.
D’autres molécules, administrées par voie systémique, peuvent être associées aux opioïdes comme les α2 agonistes, les anti-inflammatoires (stéroïdiens ou non), les antagonistes NMDA (kétamine) ou certains anesthésiques locaux (lidocaïne). C’est le concept d’analgésie multimodale.
Enfin, il est possible d’utiliser les analgésiques par voie locale. L’anesthésie locorégionale est l’une des méthodes les plus efficaces. Un bloc peut être réalisé par l’administration d’un anesthésique local (lidocaïne ou bupivacaïne) directement dans l’entourage des nerfs dont dépend le site chirurgical. L’administration péridurale des antalgiques directement au contact du système nerveux central a aussi fait la preuve de son efficacité.
Références
|
0758-1882/$ — see front matter © 2008 AFVAC. Publié par Elsevier Masson SAS. Tous droits réservés.
doi:10.1016/j.anicom.2008.09.001